Ce site est destiné aux professionnels de la santé canadiens et met à leur disposition des ressources d’apprentissage et des informations sur VEOZAH. Pour accéder au site Web sur le VEOZAH, veuillez indiquer votre profession et votre province, puis entrer votre numéro de permis d’exercice.
Toutes les marques déposées sont la propriété de leurs détenteurs respectifs.
© 2025 Astellas Pharma Inc. ou ses sociétés affiliées.
![]()
MAT-CA-VEO-2025-00007 07/25


L’efficacité de VEOZAH a été évaluée durant les 12 premières semaines de l’étude SKYLIGHT 1 et 2 de phase III, randomisée, contrôlée par placebo et à double insu. Après les 12 premières semaines, les femmes sous placebo ont été à nouveau réparties aléatoirement dans des groupes traités par VEOZAH pendant une période de prolongation du traitement de 40 semaines afin d’évaluer l’innocuité sur une durée totale d’exposition allant jusqu’à 52 semaines. Les paramètres d’évaluation principaux conjoints étaient la variation moyenne de la fréquence et de la sévérité des symptômes vasomoteurs modérés aux semaines 4 et 12 par rapport aux valeurs initiales1.
L’efficacité de VEOZAH pour le traitement des symptômes vasomoteurs modérés à sévères dus à la ménopause a été évaluée durant la première période à répartition aléatoire, contrôlée par placebo et à double insu, de 12 semaines, des deux études identiques de phase III. Lors de ces deux essais, après les 12 premières semaines, les femmes sous placebo ont été à nouveau réparties aléatoirement dans des groupes traités par VEOZAH pendant une période de prolongation du traitement de 40 semaines afin d’évaluer l’innocuité pour un total d’exposition pouvant aller jusqu’à 52 semaines1.
Paramètres d’évaluation principaux conjoints1 :
Variation moyenne de la fréquence et de la sévérité des symptômes vasomoteurs modérés à sévères aux semaines 4 et 12 par rapport au début de l’étude
Personnes participantes aux études1-3 :
Plan des études SKYLIGHT 1 et 21-3 :


RÉFÉRENCES : 1. Monographie de produit de VEOZAH. Markham (ON) : Astellas Pharma Canada Inc. 2. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102.
3. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023.
RÉFÉRENCES : 1. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102. 2. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023.
RÉFÉRENCES : 1. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102. 2. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023.


Le graphique est adapté de la monographie de produit de VEOZAH.
É.-T. : écart type; ET : erreur type.
* La supériorité par rapport au placebo est statistiquement significative au niveau de 0,05 avec ajustement pour la multiplicité1.
FRÉQUENCE : Mesurée comme une moyenne quotidienne et analysée comme une moyenne hebdomadaire (calculée comme la fréquence moyenne des symptômes sur 7 jours consécutifs)3,4.
Moyenne calculée selon la MMC : moyenne des moindres carrés estimée à partir d’un modèle mixte pour l’analyse de la covariance des mesures répétées1.


É.-T. : écart type; ET : erreur type.
* La supériorité par rapport au placebo est statistiquement significative au niveau de 0,05 avec ajustement pour la multiplicité1.
FRÉQUENCE : Mesurée comme une moyenne quotidienne et analysée comme une moyenne hebdomadaire (calculée comme la fréquence moyenne des symptômes sur 7 jours consécutifs)3,4.
Moyenne calculée selon la MMC : moyenne des moindres carrés estimée à partir d’un modèle mixte pour l’analyse de la covariance des mesures répétées1.


É.-T. : écart type; ET : erreur type.
† La supériorité par rapport au placebo est statistiquement significative au niveau de 0,05 avec ajustement pour la multiplicité1.
SÉVÉRITÉ : Mesurée en tant que moyenne quotidienne et analysée en tant que moyenne hebdomadaire3,4.
Moyenne calculée selon la MMC : moyenne des moindres carrés estimée à partir d’un modèle mixte pour l’analyse de la covariance des mesures répétées1.


É.-T. : écart type; ET : erreur type.
† La supériorité par rapport au placebo est statistiquement significative au niveau de 0,05 avec ajustement pour la multiplicité1.
SÉVÉRITÉ : Mesurée en tant que moyenne quotidienne et analysée en tant que moyenne hebdomadaire3,4.
Moyenne calculée selon la MMC : moyenne des moindres carrés estimée à partir d’un modèle mixte pour l’analyse de la covariance des mesures répétées1.


La variation moyenne de la fréquence des symptômes vasomoteurs entre le début de l’étude et chaque visite durant la phase de prolongation était un paramètre d’évaluation exploratoire. Les évaluations effectuées après la période de contrôle par placebo de 12 semaines étaient uniquement descriptives2,4.
La durée de cette étude est plus longue que celle menée pour obtenir les données sur l’efficacité incluses dans la monographie de produit de VEOZAH.


La variation moyenne de la fréquence des symptômes vasomoteurs entre le début de l’étude et chaque visite durant la phase de prolongation était un paramètre d’évaluation exploratoire. Les évaluations effectuées après la période de contrôle par placebo de 12 semaines étaient uniquement descriptives2,4.
La durée de cette étude est plus longue que celle menée pour obtenir les données sur l’efficacité incluses dans la monographie de produit de VEOZAH.


La variation moyenne de la sévérité des symptômes vasomoteurs entre le début de l’étude et chaque visite durant la phase de prolongation était un paramètre d’évaluation exploratoire. Les évaluations effectuées après la période de contrôle par placebo de 12 semaines étaient uniquement descriptives2,4.
La durée de cette étude est plus longue que celle menée pour obtenir les données sur l’efficacité incluses dans la monographie de produit de VEOZAH.


La variation moyenne de la fréquence des symptômes vasomoteurs entre le début de l’étude et chaque visite durant la phase de prolongation était un paramètre d’évaluation exploratoire. Les évaluations effectuées après la période de contrôle par placebo de 12 semaines étaient uniquement descriptives2,4.
La durée de cette étude est plus longue que celle menée pour obtenir les données sur l’efficacité incluses dans la monographie de produit de VEOZAH.
Le profil d’innocuité de VEOZAH a été évalué dans le cadre d’études cliniques de phase III
DEUX études identiques de phase III sur l’efficacité et l’innocuité, randomisées, contrôlées par placebo et menées à double insu pendant 12 semaines, suivies d’une nouvelle randomisation des femmes recevant auparavant le placebo vers VEOZAH (les femmes sous VEOZAH sont restées sous VEOZAH) pendant 40 semaines supplémentaires de traitement non contrôlé.
UNE étude de phase III de 52 semaines, randomisée, contrôlée par placebo et à double insu destinée à évaluer l’innocuité à long terme.
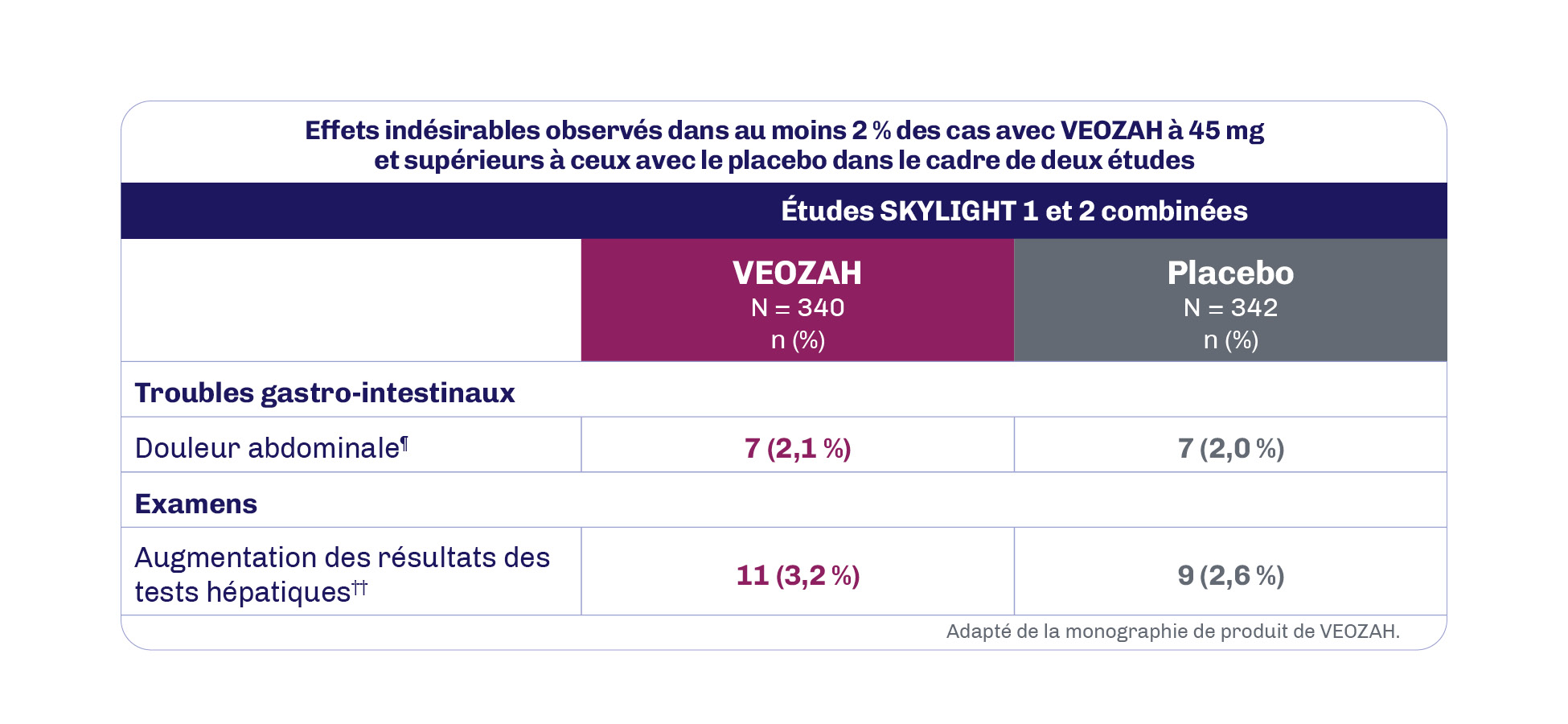
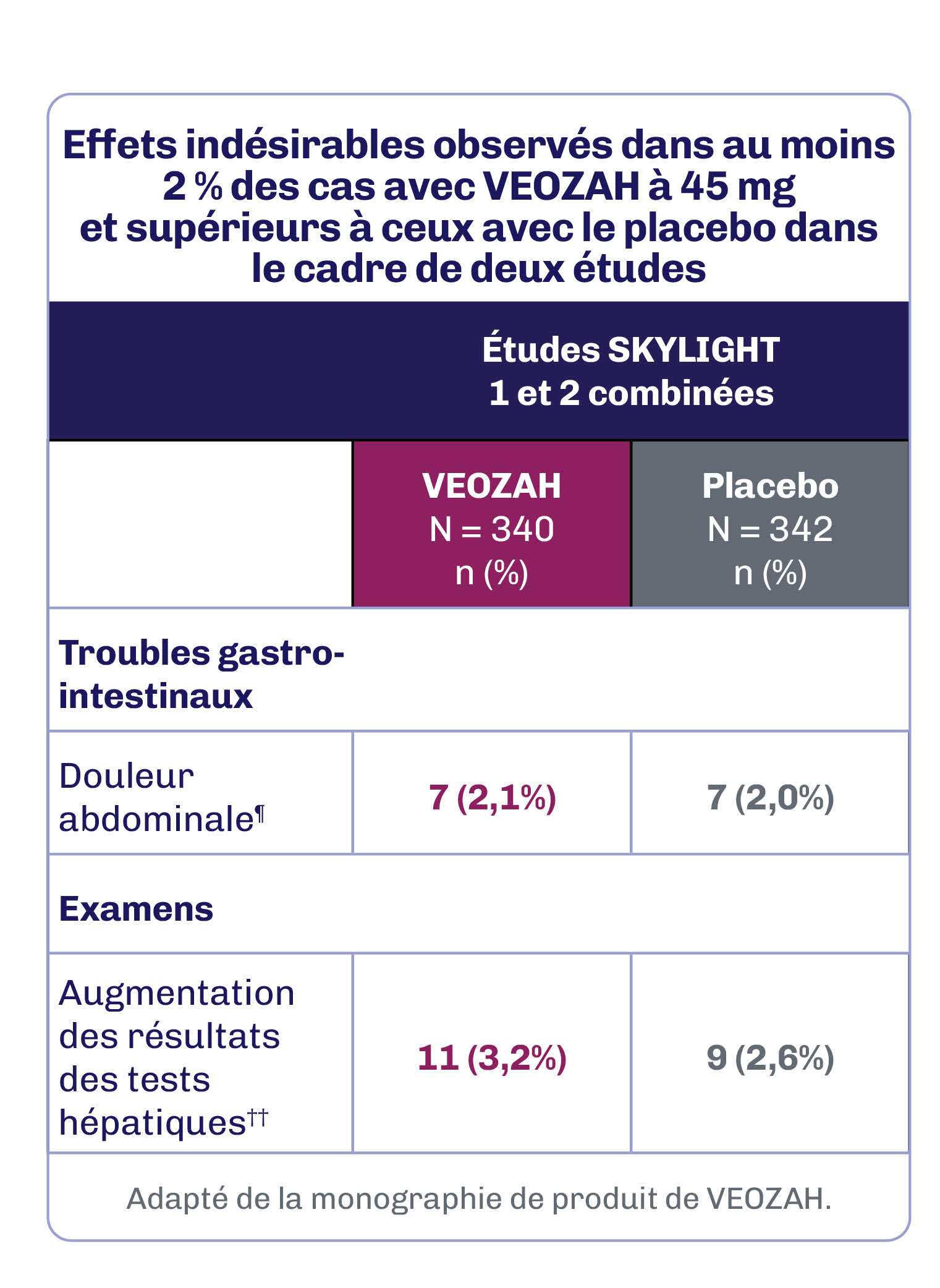
Au cours de la période contrôlée par placebo de 12 semaines des études SKYLIGHT 1 et 2, la réaction indésirable au médicament la plus fréquente (≥ 3 % chez les patientes recevant VEOZAH à 45 mg et supérieure à celle du placebo) a été l’augmentation des résultats des tests hépatiques (3,2 %)1.
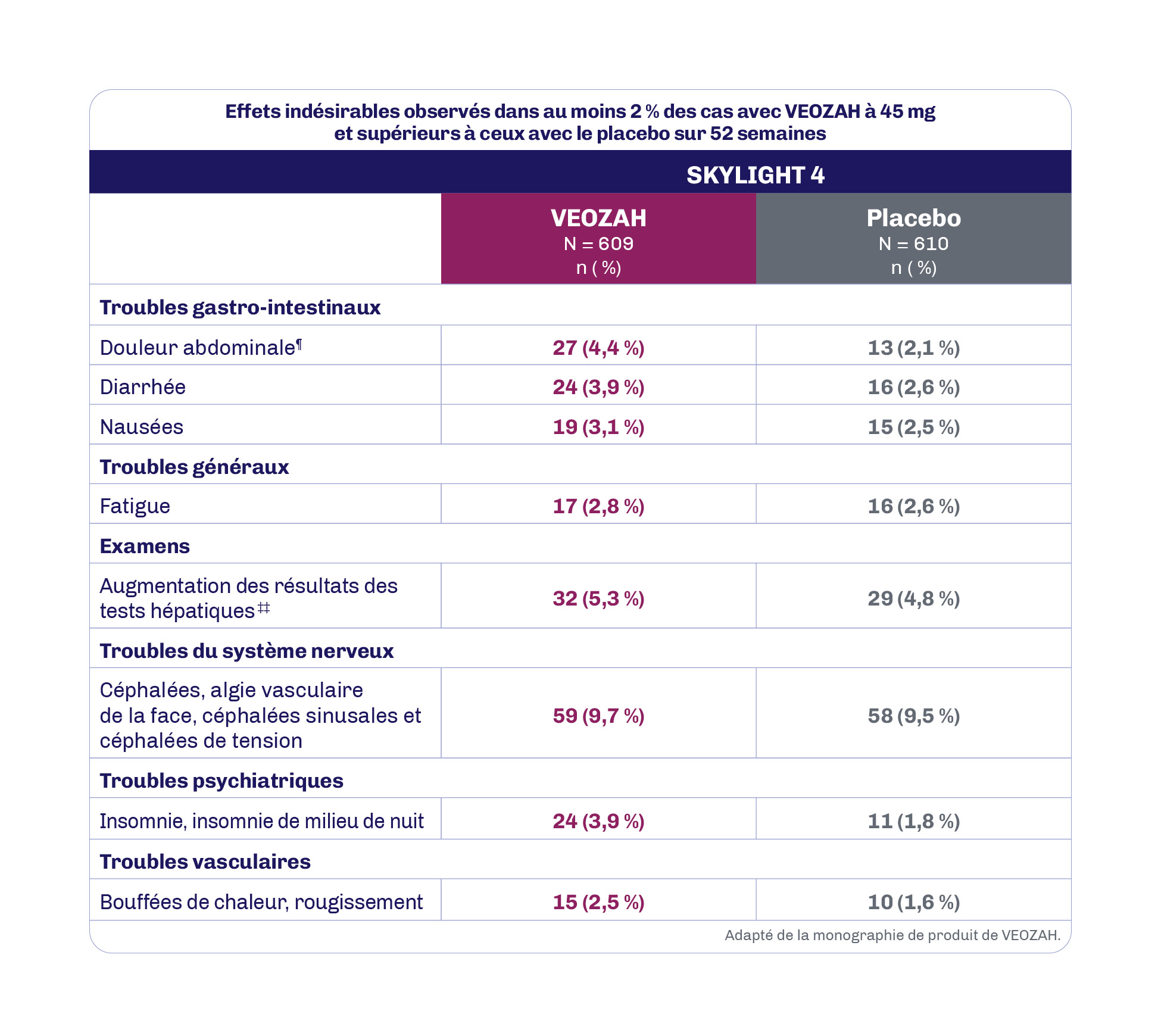
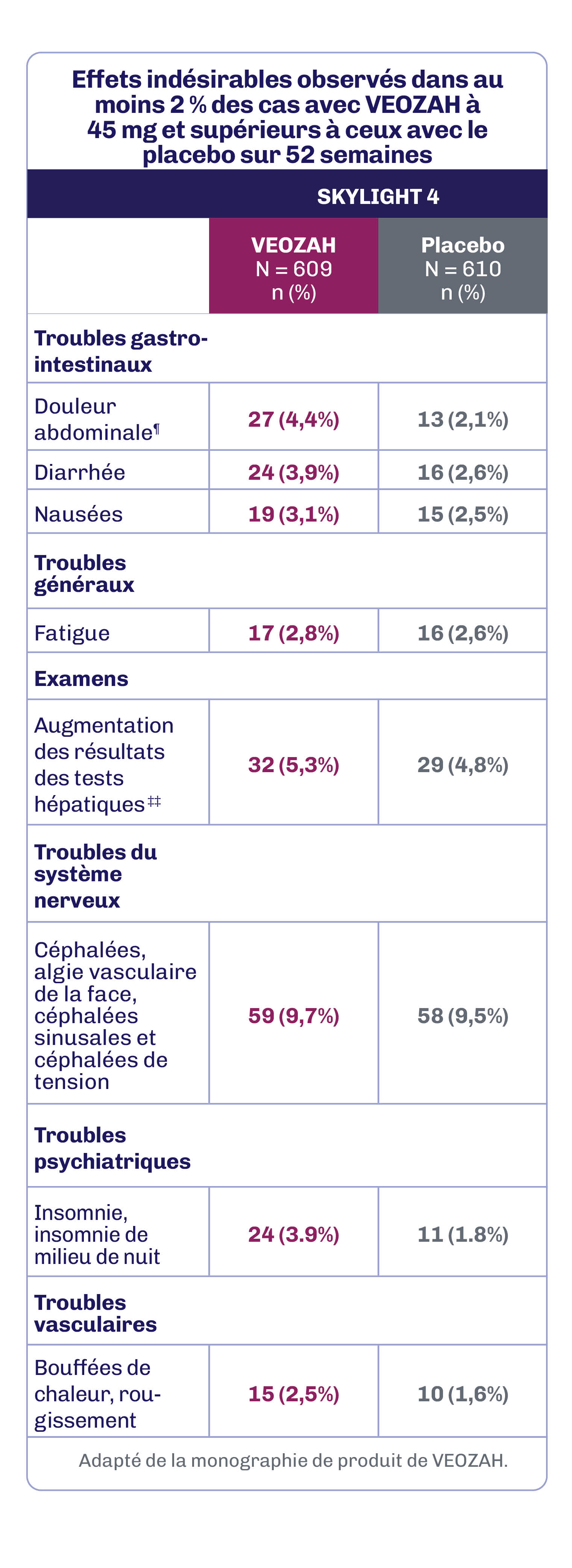
Au cours de l’étude SKYLIGHT 4 de 52 semaines contrôlée par placebo, les réactions indésirables au médicament les plus fréquentes (≥ 3 % chez les patientes recevant VEOZAH à 45 mg et supérieures au placebo) ont été les céphalées (9,7 %), l’augmentation des résultats des tests hépatiques (5,3 %), les douleurs abdominales (4,4 %), la diarrhée (3,9 %), l’insomnie (3,9 %) et les nausées (3,1 %)1.
Un déséquilibre numérique a été observé dans l’incidence des autres tumeurs malignes entre le groupe VEOZAH et le groupe placebo dans l’étude sur l’innocuité à long terme (SKYLIGHT 4). Une relation de cause à effet entre VEOZAH et l’augmentation du risque de tumeurs malignes n’a pas été établie1.
L’étude SKYLIGHT 4 était une étude de phase III d’une durée de 52 semaines à répartition aléatoire, contrôlée par placebo et à double insu évaluant l’innocuité à long terme chez des femmes présentant des symptômes vasomoteurs associés à la ménopause1.
Paramètres d’évaluation principaux2:

Caractéristiques démographiques et initiales2:
L’âge moyen des femmes ayant atteint la ménopause était de 55 ans. Les femmes se sont identifiées comme blanches (79,9 %), noires ou afro-américaines (17,2 %) et asiatiques (1,6 %), et appartenant à plus d’une race (0,9 %). L’origine ethnique a été définie comme hispanique/latino-américaine (20,1 %) ou non hispanique/latino-américaine (79,9 %). La population de l’étude comprend des femmes atteignant la ménopause qui ont des antécédents d’hormonothérapie (17,0 %) et qui ont subi une ovariectomie (13,5 %) ou une hystérectomie (18,6 %). L’indice de masse corporelle était de 28,4 ± 4,6. Le pourcentage de fumeuses était de 19,1%.
RÉFÉRENCES : 1. Monographie de produit de VEOZAH. Markham (ON) : Astellas Pharma Canada, Inc. 2. Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141(4):737-47.
RÉFÉRENCE : 1. Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141(4):737-47.
RÉFÉRENCE : 1. Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141(4):737-47.
ALT : alanine aminotransférase; AST : aspartate aminotransférase; PA : phosphatase alcaline.
RÉFÉRENCES : 1. Monographie de produit de VEOZAH. Markham (ON) : Astellas Pharma Canada, Inc. 2. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401(10382):1091-102. 3. Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. (Epub) 02-03-2023. 4. Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study (annexe supplémentaire). Lancet. 2023;401(10382):1-14. 5. Données internes. Résumé sur l’efficacité clinique.